Medical devices – Quality management systems –
Requirements for regulatory purposes
의료기기의 GMP(Good Manufacturing Practice) 국제 규격인 ISO 13485는 1996년 제정되어 2003년 2차 개정을 통해 2016년 3월 1일 3차(Third edition) 개정되었다.
3년의 유예기간이 만료되는 2019년2월28일까지는 Third edition으로 인증 전환이 완료되어야 한다.
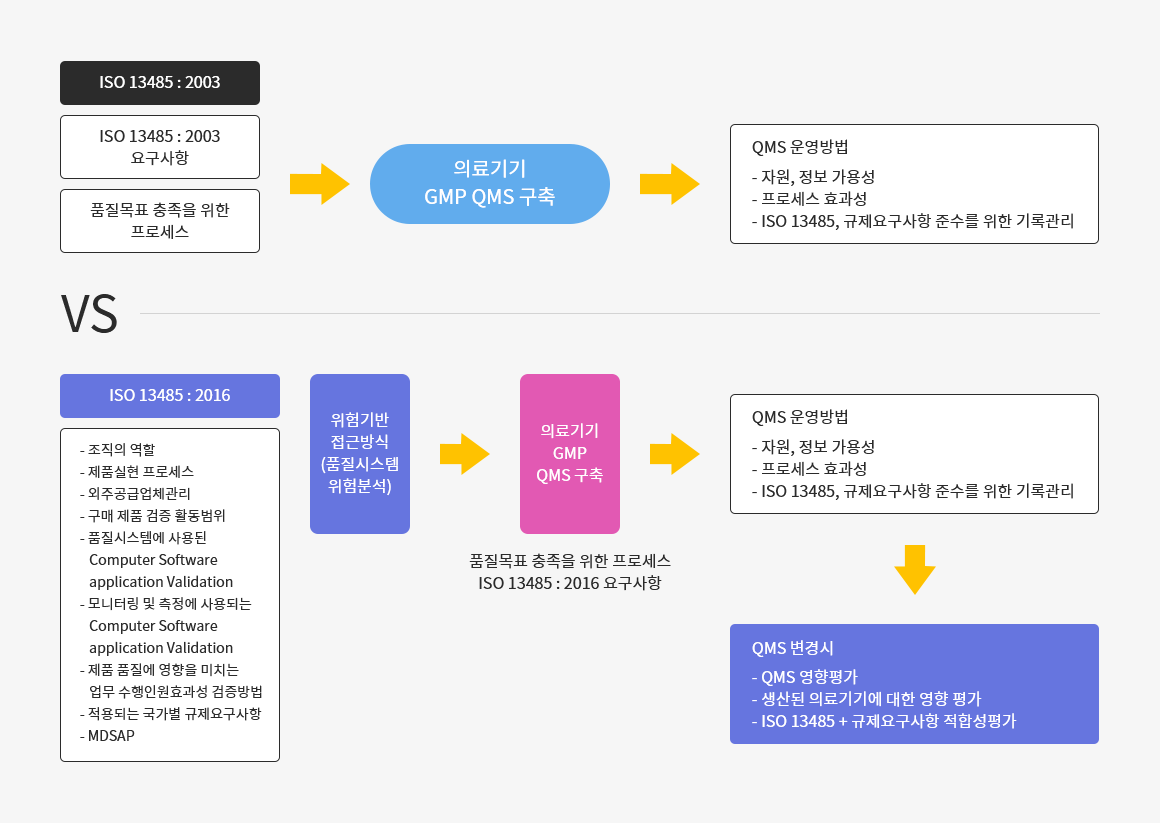
Third edition의 주요 개정사항으로 조직의 역할을 문서화 하도록 요구함으로써 의료기기 제조사뿐만 아니라 의료기기 life-cycle에 참여하는 모든 조직으로 ISO 13485 적용 조직을 확대하였으며,조직에 적용되는 규제 요구사항을 파악하여 적용하도록 요구하고 있다.
또한, 품질시스템 구축 시 위험에 비례하여 관리를 요구함으로써 품질시스템에 대한 위험분석을 통해 품질시스템 구축을 요구한다.
주요 개정사항을 구체적으로 살펴보면 다음과 같다.
01. ISO 13485 인증 조직 범위 확대
- ISO 13485:2003에서는 조직(의료기기 제조자)에 대하여 적용
- - 의료기기 제조사에 한정하여 적용
- ISO 13485:2016에서는 조직이 맡은 역할을 문서화 하도록 요구
- - 의료기기 life-cycle에 참여하는 모든 조직으로 범위가 확대
* ISO 13485 인증: 의료기기 제조사, 외주공급업체, 운송업체, 수입업체, 유통업체, 설치업체, 수리업체, 위임대리인 등
- - 의료기기 life-cycle에 참여하는 모든 조직으로 범위가 확대
02. 품질시스템 위험분석
ISO 13485:2003 vs ISO 13485:2016 품질경영시스템 구축 프로세스 개념
03. 적용되는 규제 요구사항을 31개 항에서 요구
규제 요구사항을 반영한 품질시스템 구축
- - 대한민국 : 의료기기법, 시행령, 시행규칙, 고시 및 규정, 의료기기 제조 및 품질관리 기준
- - 유럽 : Directive(90/385/EEC, 98/79/EC, 93/42/EEC), EN ISO 13485, MEDDEV, PMS, RoHS, WEEE etc
- - MDSAP :

- 규제 요구사항 준수를 위한 기록관리
- 설계 입력 및 변경 시 규제 요구사항 포함
- 규제 요구사항에 따른 UDI 시스템 문서화
- 규제 요구사항에 따른 불만 처리 절차 문서화
- 시정 및 예방조치 시 규제 요구사항에 부정적 영향 검증
04. 위험에 비례하여 관리 요구
- 외주업체 관리 : 외주업체별 위험에 비례한 선정평가, 품질합의서, 재평가 시스템
- 품질시스템에 사용되는 컴퓨터 소프트웨어 어플리케이션 Validation
- 생산 및 서비스 제공에 사용되는 컴퓨터 소프트웨어 Validation
- 모니터링 및 측정에 사용되는 컴퓨터 소프트웨어 어플리케이션 Validation
05. 설계검증 및 유효성 확인 계획 요구
개발 스펙 별 검증/유효성 확인 계획 수립을 통해 추적성 확보를 요구한다.
