Quality Systems Regulation
미국의 의료기기의 cGMP(current Good Manufacturing Practice) 법률인 21 CFR PART 820-QSR은 1978년 최초 제정되어 현재까지 개정되며 적용되고 있다.
미국 cGMP는 별도의 인증제도가 없으며, FDA에 의료기기를 등록한 제조사는 21 CFR PART 820-QSR에 따라 품질시스템을 수립하고 유지하여야 한다.
cGMP Inspection은 FDA CDRH(Center for Devices and Radiological)에서 주관하고 있으며, CDRH의 업무 범위는 의료기기, 엑스레이, 레이저, 초음파, 마이크로웨이브 등의 방사선 노출 전자기기에 대하여 미국 내에서 판매되는 제품의 제조, 포장, 라벨, 수입하는 회사를 관리한다.
cGMP Inspection은 CDRH에서 배정하며 통상 2~3개월 전에 FDA 위임 대리인(designated Agent)을 통해 통보가 된다. 또한 제조사는 품질매뉴얼 영문본을 제출해야 한다. (일정 통보 > 제조사 일정 의견 제출 > 일정 확정 > 심사원(Inspector) 배정 및 제조사로 연락)
01. 심사(Inspection) 대상
- FDA에 등록된 specification developer, manufacturer & specification developer
- Class 1 : 원칙적으로 Inspection 대상 제외 (단, 건강 위험성이 명백하거나, 특별한 사유가 발생할 경우 Inspection 배정)
- Class 2,3 : CDRH에서 Inspection 배정(위험에 기반하여 배정(우선순위))
- PMA(Premarket Approval)심사대상인 제조자
- 심사가 이루어지지 않은 Class 3 제조자
- 부적합 사항에 대한 후속 실사 또는 특정사안이 있는 경우에만 실사
- 리스크가 높은 의료기기 제조자로
- - CDRH의 특별배정 업체
- - 리콜과 MDR(Medical Device Reporting) 빈도가 높은 의료기기
- - 소프트웨어가 장착되고 기술변화가 빠르게 진행되어 이러한 변화가 안전성과 성능에 영향을 미칠 수 있는 의료기기 제조업체
- - 신개발 의료기기
- 일회용기기 재 멸균업자, 재 멸균병원, 계약 멸균업자
02. 심사(Inspection) 단계
| 심사(Inspection)단계 심사(Inspection) Level |
심사(Inspection)형태 심사(Inspection) Type |
심사(Inspection)의 사유 심사(Inspection) Reason |
|---|---|---|
| 1단계 Level 1 | 약식 Inspection Abbreviated |
일방적인 심사-문제가 없는 경우 Normal-No problems |
| 2단계 Level 2 미국 외 해외 Inspection는 2단계에서 시작 |
포괄 Inspection Comprehesnsive |
최초 FDA Inspection 또는이전 Inspection 후 오랜 기간이 지났을 때 No prior history, long time since last one |
| 3단계 Level 3 |
직접여부확인후속 Inspection Comprehesnsive Flow-up |
이전 Inspection에서 경고장을 받은경우 또는 이전 Inspection에서 부적합사항이 있는 경우 After Warning Letter or other finding |
| 특별 Inspection Special | 특정시안 For Cause |
시장, 생산시설 등에서 문제가 발생된 경우 Problems-in field, in facillity |
cGMP Inspection 제외 대상
- - 일반적으로 Class 1 제조자
- - 계약수리업자, 재처리업자, 서비스제공자, 중고의료기기 재판매업자
- - 일회용 의료기기 재 멸균병원은 CDRH의 특별요청이 있을 경우에만 실사
03. 심사(Inspection) 일정 및 프로세스
- Inspection 일정 : 4일간 수행
- 품질시스템(QMS/cGMP Regulation) Inspection 프로세스
Main process
- 1일차 : Management Control(경영관리) : 820.20, 22, 25
- 2일차 : Design Control(설계관리) : 820.30
- 3일차 : Corrective and Preventive Actions(CAPA)(시정 및 예방조치) : 820.100
- - 의료기기 사고보고(MDR Regulation) : 820, 803
- - 제품 수리 및 회수(Recall Regulation) : 820, 810
- - 추적관리 (Tracking requirements) : 820.65
- 4일차 : Production and Process Controls(P&PC)(생산 및 공정관리)
: 820.50, 60, 70, 72, 75, 80, 86, 90, 120, 130, 150, 170, 200, 250
Sub process
- - Facilities and Controls(시설 및 장비관리) : 820. 70(f, g)
- - Material Controls(자재관리) : 820.140, 150
- - Documents/Records/Change Control(문서/기록/변경 관리) : 820.40, 180, 181, 184, 186, 198
- 제품등록(Registration and Listing Regulation) : 820.807
04. 심사(Inspection) 지적사항 type
FDA Inspector는 Inspection을 수행 후 지적사항을 FDA 483 Observations 또는 Warning Letter로 발행한다. Inspection을 수검 받은 제조사는 조치 계획을 수립한다. 제조사는 이 사항을 Inspector 및 CDRH에 메일로 제출하여 검토를 받고 지적사항에 대한 후속조치를 수행한다.
Inspection 지적사항에 따라 Warning Letter를 받을 수 있다. 이는 미국내 의료기기 반입을 금지하는 출하중지 명령을 포함할 수도 있다.
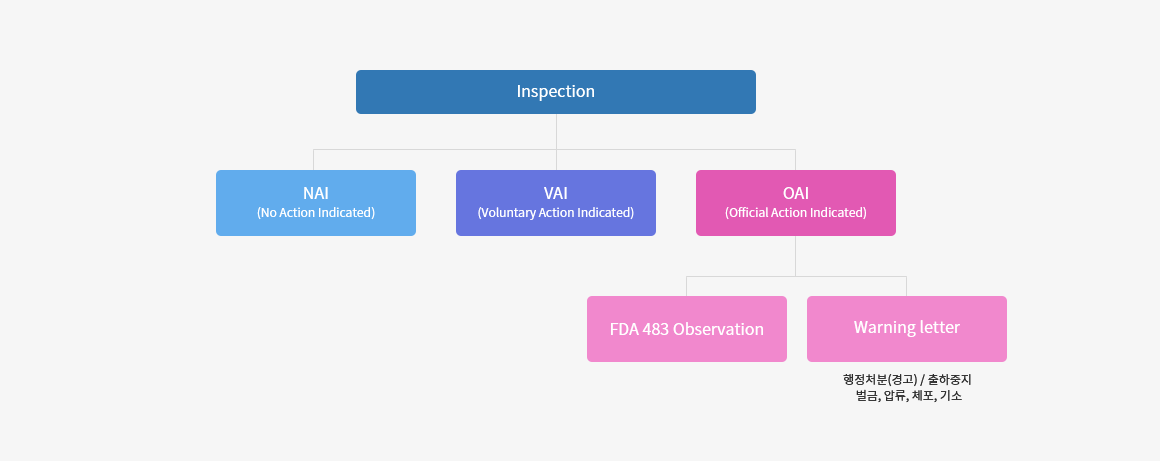
05. 심사(Inspection) 결정형태
- No Action Indicated(NAI) : 위해 행위가 발견되지 않음
- Voluntary Action Indicated (VAI) : 지적사항은 아니나 수정사항을 담당자에게 통보
- Official Action Indicated (OAI) : 지적사항으로 법적, 행정적, 사법적 조치가 권고되는 상태(자발적 리콜도 해당됨)
- - 심각한 건강위험성이 파악되고 회사에서 자발적 리콜을 수행하지 않은 경우 :
강제 리콜, 구속, 영업정지(출하중지 명령), 행정처분(Warning Letter(경고), Injunction(중지명령), Detention(구금),
Seizure(압류), Civil penalty(벌금), Prosecution(기소) 등 - - 품질시스템 결함에 대한 미비한 가능성만 있을 경우 :
483 Inspection Observations 발행
- - 심각한 건강위험성이 파악되고 회사에서 자발적 리콜을 수행하지 않은 경우 :